

主页 > 检测中心 > 脆性X综合征基因筛查
脆性X综合征基因筛查

在Xq27.3处有脆性部位的X染色体称为脆性X染色体(fragile X),与之相关的疾病称为脆性X综合征(fragile X syndrome,FXS)[OMM 309550],又称Martin-Bell综合征,是最常见的X连锁隐性遗传的单基因性智力低下综合征,占所有X-连锁智力低下疾病的15-25%,发病率仅低于21-三体综合征。超过99%的脆性X综合征患者是由FMR1基因三核苷酸重复顺序扩增和超甲基化所致,少于1%的患者因FMR1基因部分缺失或点突变而致病。该病在世界各个人群中都有发现,发病者多为男性,危害很大。

● 导致智力低下
● 生殖系统发育异常
● 脸面部发育异常
● 神经系统疾病
● 语言能力障碍
● 数学学习能力障碍
● 神精疾病
● 多系统并发症
● 严重降低生存质量
● 给家庭、社会带来巨大负担

女性群体中FMR1前突变携带率为1/130-1/256,携带者有50%的几率将FMR1突变基因遗传给下一代,其外显率因性别不同有差异,男性80-100%,女性30-40%。本病在男性群体中发病率为1:4000,女性群体中发病率为1:6000,FXS在所有智力低下病例中约占2-4%。

FMR1基因是脆性X综合征的致病基因,位于Xq27.3,基因长38kb,含17个外显子。外显子之间的拼接方式有多种,从而产生不同的mRNA,可翻译出不同的蛋白质亚型。脆性X染色体智力迟钝蛋白(fragile X mental retardation protein,FMRP)是 FMR1基因翻译产物,它是一种RNA结合蛋白,其结构中含有能结合RNA的功能区段,同时还有细胞核输出讯号区段(nuclear export signal,NES)和细胞核定位讯号区段(nuclear location signal,NLS)。所以FMRP的功能是和特定的mRNA结合,并将之从细胞核内携带转运至特定的细胞浆亚区进行翻译。FMRP则可以在细胞核和胞浆之间来回穿梭,被认为能够调控神经发育期间特定基因的表达,影响神经细胞突出的可塑性。FMRP对大脑的发育很重要,在脑的一些部分,如尾叶、丘脑、小脑、海马、颞回合大脑皮质,有很高的表达。FMRP的缺乏对大脑的发育和功能影响很大,如何造成智力和行为方面的临床症状的更深入病理机制还在研究之中。超过99%的脆性X综合征的患者是由FMR1三核苷酸重复顺序扩增(CGG)n和超甲基化所致,少于1%的患者因FMR1基因部分缺失或点突变而致病,属于X连锁隐性遗传。
● (CGG)n重复顺序的扩增程度可以分为:
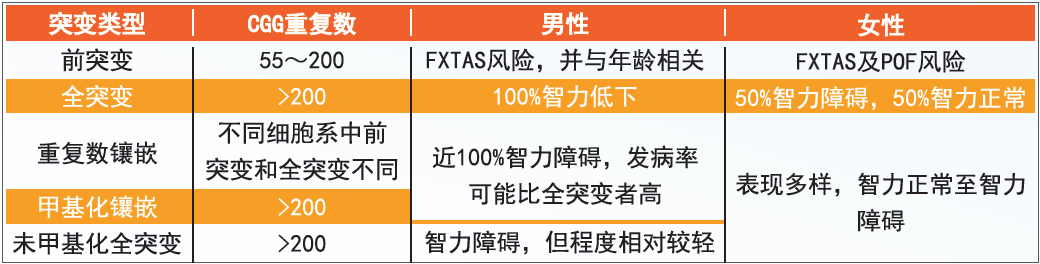
前突变(premutation):(CGG)n重复顺序在55-200之间,通常无CpG岛的超甲基化。前突变的携带者FMR1基因的转录活跃,产生比正常人高的mRNA,但其翻译效率低,故FMRP产量比正常人的低。过去认为,前突变的携带者为无临床症状的正常人,但能将可扩增的基因传递给下一代;现在认为,大量FMR1 mRNA会和一些蛋白质结合,在神经细胞中形成包涵体导致神经细胞功能异常甚至死亡。CGG重复数为45-54的女性传递给后代成前突变的概率为4.7%,传递给后代成为全突变的概率为0;CGG重复数为55-199的女性传递给后代为前突变的概率为41.2%,全突变的概率为58.8%。
全突变(full mutation):重复顺序超过200个,见于男性、女性患者和女性携带者。扩增后(CGG)n区和FMR1基因的启动子CpG岛出现超甲基化(hypermethylation)。高度扩增的重复顺序和超甲基化抑制了FMR1基因的表达,导致FMRP缺乏。
镶嵌性突变(mosaicism):约15-20%的脆性X综合征患者有镶嵌性突变。有两种镶嵌状态,一种是指患者的体细胞中同时具有前突变与全突变,即重复顺序长度的镶嵌;另一种是患者的体细胞中同时具有超甲基化和非甲基化的FMR1 CpG岛,即甲基化的镶嵌。这两种镶嵌状态可以单独或同时出现,一般来说,具镶嵌状态的患者智力低下的程度较轻。
● 孕产前女性FMR1基因DGG重复数与胎儿疾病风险

● 新生儿FMR1基因CGG重复数与疾病风险
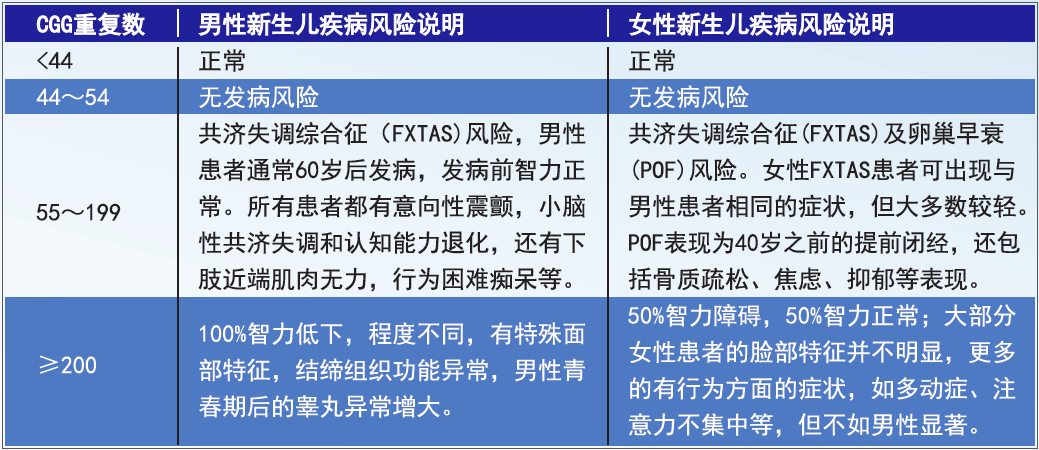
● 男性、女性群体中FMR1基因CGG重复数突变类型与疾病风险

脆性X 综合征是一种家族性智力障碍疾病,临床以智力低下、特殊面容、巨睾症、大耳、语言和行为异常为其典型表现。并有诸多并发症:智力发育迟滞、生长障碍、精神心理障碍、躁狂、癫痫、内耳感染(中耳炎)、视力问题(包括近视、眼球震颤)以及消化系统紊乱(包括胃食管反流等)等。
● 男性患者典型临床症状
特殊面容:长脸,有时前额突出,高腭弓,厚唇,大耳朵,单耳轮,大下巴,下颌前突。出生体重多较高,生后头几年生长速度快,但成人时身材矮小。
大睾丸:青春后期80%患者出现该症状。多在青春前期睾丸增大,睾丸较正常人大2-6倍。阴囊增厚,少见于年幼患者,常伴大阴茎。
语言发育障碍:较为常见,表现为会话和言语表达能力的发育严重迟缓,存在构音障碍、病理性模仿和重复言语以及语法和词汇缺乏等。
精神发育障碍:有学习能力障碍、注意力集中障碍、情绪问题(如焦虑、沮丧、害羞等)以及社会交往能力低下。
人格行为异常:带有孤独症的特征。患儿易怒,注意力集中困难,高度焦虑,并容易对周围发生的事情歇斯底里。
神经系统症状:多较轻微,常见为四肢运动障碍。不随意运动迟缓、关节过度强直及全身反射亢进,偶见抽风等,20%患者有癫痫发作。
生殖系统:性功能低下,成年患者部分器官女性化,但可生育后代。
其他:80%患者有扁脚,成年期50-80%患者出现二尖瓣脱垂,60%有复发性中耳炎,30%可出现斜视,20%有屈光不正,15%出现痉挛发作,小于20%有脊柱侧弯。
● 女性携带者临床症状
可出现轻度智力低下。在女性携带者中,70%有前突变而不表现出明显的生理或认知、行为异常;其他30%具有全突变的女性,会表现一定范围的症状。
外观表现:一般发病女性在外貌上只有轻微的改变,有尖脸和大耳,异常身体特征较少。只有1/3-1/2的女性患者表现出这些特征。
数学学习能力障碍:全突变的女性认知和行为方面的异常,通常会比在发病男性身上所观察到的症状轻。有一半表现出典型的智力低下和表现出非言语性学习困难,另一半有通常需要得到不断支持的精神发育迟滞。发病女性在词语记忆和阅读方面有近似的能力,数学学习能力差,也有一种特别的说话方式,表现为重复和杂乱无章。
注意力集中障碍:刻板行为,离题的说话,冲动,注意力分散和适应困难。
情绪问题: 全突变的女性中有 20%~60%患有精神心理障碍。最常见抑郁,特有的精神分裂样改变,广泛发育障碍,个性退缩和焦虑。
社会交往能力低下:个性害羞和退缩,有奇怪的交流方式和怪癖。

天昊诊断使用特殊荧光PCR方法(美国Amplide X检测技术,已获欧洲CE-IVD认证)检测FXS致病基因FMR1基因的CGG重复数,本检测快速准确,只需要少量基因组DNA,针对致病DNA片段区域设计特异检测荧光探针,通过特殊荧光PCR扩增,再经毛细管电泳检测及生物信息学分析,即可精确估算FMR1基因的CGG三核苷酸串联重复的长度。
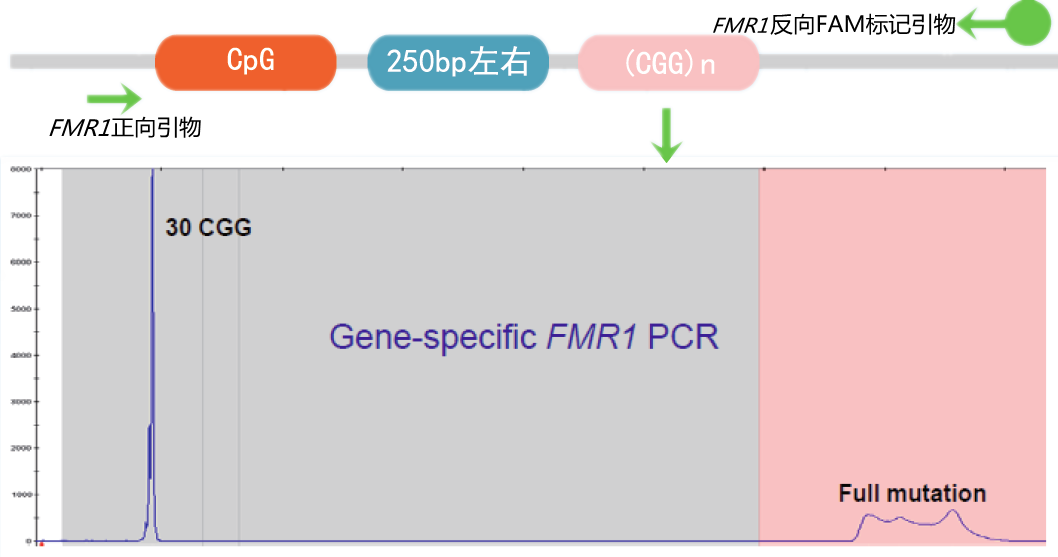
本方法简单、高效、准确,与欧美国家及地区的FXS筛查方法相同,与其他检测方法相比有明显优势。


正常人群中妇女FXS携带率极高,携带者将此病传递给下一代的概率高,孕产前妇女筛查FXS可以最大程度避免患儿出生,新生儿筛查可以使结果阳性者得到及时正确的治疗,利于优生优育。
备孕女性:了解自身FMR1 CGG 突变状态,确定是否为携带者,评估孕育风险,制定合理的孕育计划。对于前突变以上的一定要做好产前诊断,对于CGG重复数为50-54的由于有10%的前突变发生,也建议做好产前诊断。
妊娠小于4个月的准妈妈:了解自身FMR1 CGG 突变状态,高危孕妇可及时采取有效预防措施。全突变携带者会有50%概率传递给后代男性,而且会使得后代女性也为携带者,前突变携带者在卵子产生过程中很容易发生突变,产生全突变卵子使得后代为FXS患儿或全突变携带者。
新生儿:前突变以上均有高风险,及时发现FXS患病风险或突变携带状态,特别是女孩携带者,以便尽早干预和治疗。
临床可疑患者:携带者筛查,确定FMR1 CGG突变情况,辅助临床确诊。



样本类型:EDTA或枸橼酸钠抗凝(避免用肝素抗凝)外周血1ml,或唾液2ml,或口腔拭子2根。
保存及运输:2-8℃可存放1周,更长时间采用-20℃保存;运输时需用泡沫箱加冰袋密封,运输途中避免暴晒且时限不超过72小时。

1. Hagerman, Randi Jenssen, and Paul J. Hagerman, eds. Fragile X syndrome: Diagnosis, treatment, and research. JHU Press, 2002.
2. Fu Y H, Kuhl D, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox[J]. Cell, 1991, 67(6): 1047-1058.
3. McConkie-Rosell A, Finucane B, Cronister A, et al. Genetic counseling for fragile X syndrome: updated recommendations of the National Society of Genetic Counselors[J]. Journal of genetic counseling, 2005, 14(4): 249-270.
4. Sherman S, Pletcher B A, Driscoll D A. Fragile X syndrome: diagnostic and carrier testing[J]. Genetics in Medicine, 2005, 7(8): 584-587.
5. Verkerk A J M H, Pieretti M, Sutcliffe J S, et al. Identification of a gene (FMR1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome[J]. Cell, 1991, 65(5): 905-914.
6. Cornish K, Kogan C, Turk J, et al. The emerging fragile X premutation phenotype: evidence from the domain of social cognition[J]. Brain and cognition, 2005, 57(1): 53-60.
7. Goodlin-Jones B L, Tassone F, Gane L W, et al. Autistic spectrum disorder and the fragile X premutation[J]. Journal of Developmental & Behavioral Pediatrics, 2004, 25(6): 392-398.
8. Santoro M R, Bray S M, Warren S T. Molecular mechanisms of fragile X syndrome: a twenty-year perspective[J]. Annual Review of Pathology: Mechanisms of Disease, 2012, 7: 219-245.
9. Peprah E. Fragile X syndrome: the FMR1 CGG repeat distribution among world populations[J]. Annals of human genetics, 2012, 76(2): 178-191.